News
People currently working in our group:






























HIOS
 Optoelectronic excitations at inorganic/organic interfaces:
Optoelectronic excitations at inorganic/organic interfaces:
Role of vibrations and time scales

This project (2013-2019) was dedicated to the ab initio description of temperature-dependent exciton spectra of hybrid inorganic/organic semiconductor interfaces by a combination of density-functional theory (DFT) with many-body perturbation theory. Target systems included single molecules, mono- and multilayers of functionalized molecules like pyridine, oligo-acenes, -thiophenes and -phenylenes adsorbed on ZnO, Si, and GaN surfaces.
Recent publications:
|
W. Olovsson, T. Mizoguchi, M. Magnuson, S. Kontur, O. Hellman, I. Tanaka, and C. Draxl With the examples of the C K-edge in graphite and the B K-edge in hexagonal BN, we demonstrate the impact of vibrational coupling and lattice distortions on the X-ray absorption near-edge structure (XANES) in 2D layered materials. Theoretical XANES spectra are obtained by solving the Bethe-Salpeter equation of many-body perturbation theory, including excitonic effects through the correlated motion of core-hole and excited electron. We show that accounting for zero-point motion is important for the interpretation and understanding of the measured X-ray absorption fine structure in both materials, in particular for describing the σ*-peak structure. Read more... |
|
|
P. Garcia Risueno, D. Nabok, Q. Fu, and C. Draxl We present a novel approach to electron-lattice interaction beyond the linear-coupling regime. Based on the solution of a Holstein-Peierls-type model, we derive explicit analytical expressions for the eigenvalue spectrum of the Hamiltonian, resulting in a narrowing of bands as a function of temperature. Our approach enables the intuitive interpretation in terms of quasiparticles, i.e. polaron bands and dressed-phonon frequencies. Being nonperturbative, the formalism also applies in the strong-coupling case. We apply it to the organic crystal naphthalene, with the coupling strengths obtained by ab initio calculations. Read more... |
|
 |
Beyer, D. Pham, C. Peter, N. Koch, L. Grubert, S. Hecht, D. Nabok, C. Cocchi, C. Draxl, and A. Opitz Controlling the electrical conductivity of organic semiconductors is a key asset for organic electronics, nowadays realized mostly by molecular dopants. Two doping mechanisms have been reported – charge-transfer complex (CTC) and ion pair (IPA) formation. However, their occurrence depending on molecular structure, energy levels, and structure of thin films remains elusive. Here, we study p-type doping of the planar organic semiconductor dibenzotetrathiafulvalene (DBTTF) in combination with the electron acceptors tetracyanonaphthoquinodimethane (TCNNQ) and hexafluorotetracyanonaphtho-quinodimethane (F6TCNNQ) as planar dopants.We identified two different CTCs for DBTTF:TCNNQ blends and both types of charge-transfer interactions (CTC and IPA) in films of DBTTF doped with F6TCNNQ from absorption measurements. No signature of the charge-transfer interaction is found for DBTTF and TCNNQ in solution, whereas IPA formation only is observed for DBTTF and F6TCNNQ. Many-body perturbation theory complements the experimental data and helps in understanding the behavior of CTCs ... Read more... |
|
Q. Fu and C. Draxl Single-atom catalysts (SACs) combine the best of two worlds by bridging heterogeneous and homogeneous catalysis. The superior catalytic properties of SACs, however, can hardly be exploited without a suitable substrate. Here, we explore the possibility of using hybrid organic-inorganic perovskites as supporting materials for single transition-metal atoms. By means of first-principles calculations, we predict that single Pt atoms can be incorporated into methylammonium lead iodide surfaces by replacing the methylammonium groups at the outermost layer. The iodide anions at the surface provide potentially uniform anchoring sites for the Pt atoms and donate electrons, generating negatively charged Pt1δ− species that allow for preferential O2 adsorption in the presence of CO. Such Pt sites are able to catalyze CO oxidation and may also play a role in CO2 reduction. The fundamental understanding generated here will shed light on potential applications of hybrid perovskites in the field of (photo)catalysis. Read more... |
|
 |
C. Cocchi, T. Breuer, G. Witte, and C. Draxl Pentacene is one of the most studied organic materials, still a number of issues are debated. This concerns, for instance the role of polymorphism and how it affects the lowest-energy exciton, which appears in the visible region and is subject to a sizable Davydov splitting. We address this problem in a combined theoretical and experimental work, where the optical absorption properties of three pentacene polymorphs are investigated within the whole energy range of visible light. Optical spectra computed from first principles in the framework of many-body perturbation theory are directly compared with the polarization-resolved absorbance, measured for three different pentacene phases (the two bulk polymorphs and the thin-film phase). In this way, we unambiguously identify the two Davydov components of the first exciton and the optical fingerprints of each considered phase. With very good agreement between theory and experiment, we show that all polymorphs exhibit common features at the absorption onset, while phase-dependent characteristics appear only above 2 eV. We discuss the character of the lowest-lying singlet and triplet excitons, including dark ones, highlighting the contributions from the electronic bands and the role of the electron–hole interaction and of the local-field effects. Read more... |
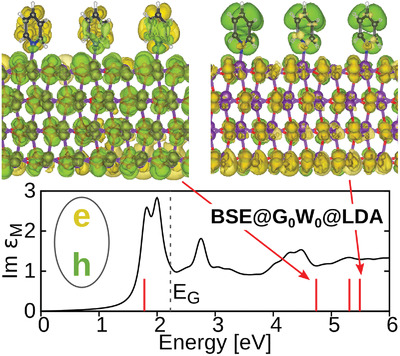 |
O. Turkina, D. Nabok, A. Gulans, C. Cocchi, and C. Draxl By combining all‐electron density‐functional theory with many‐body perturbation theory, a prototypical inorganic/organic hybrid system, composed of pyridine molecules that are chemisorbed on the nonpolar ZnO( 101¯0) surface is investigated. The G0W0 approximation is employed to describe its one‐particle excitations in terms of the quasiparticle band structure, and the Bethe–Salpeter equation is solved to obtain the absorption spectrum. The different character of the constituents leads to very diverse self‐energy corrections of individual Kohn–Sham states, and thus the G0W0 band structure is distinctively different from its DFT counterpart, that is, many‐body effects cannot be regarded as a rigid shift of the conduction bands. The nature of the optical excitations at the interface over a wide energy range is explored and it is shown that various kinds of electron‐hole pairs are formed, comprising hybrid excitons and (hybrid) charge‐transfer excitations. The absorption onset is characterized by a strongly bound bright ZnO‐dominated hybrid exciton. For the selected examples of either exciton type, the individual contributions from the valence and conduction bands are analyzed and the binding strength and extension of the electron‐hole wavefunctions are discussed. Read more... |
 |
L. Pithan, D. Nabok, C. Cocchi, P. Beyer, G. Duva, J. Simbrunner, J. Rawle, C. Nicklin, P. Schäfer, C. Draxl, F. Schreiber, and S. Kowarik We present a combined experimental and theoretical study to solve the unit-cell and molecular arrangement of the tetracene thin film (TF) phase. TF phases, also known as substrate induced phases (SIPs), are polymorphs that exist at interfaces and decisively impact the functionality of organic thin films, e.g., in a transistor channel, but also change the optical spectra due to the different molecular packing. As SIPs only exist in textured ultrathin films, their structure determination remains challenging compared to bulk materials. Here, we use grazing incidence X-ray diffraction and atomistic simulations to extract the TF unit-cell parameters of tetracene together with the atomic positions within the unit-cell. Read more... |
 |
R. R. Pela, A. Gulans, and C. Draxl The LDA-1/2 method evaluates ionization potentials as the energy of the highest occupied molecular orbitals (HOMOs) at half occupation. It has proven to be a viable approach for calculating band gaps of semiconductors. To address its accuracy for finite systems, we apply LDA-1/2 to the atoms and molecules of the GW100 test set. The obtained HOMO energies are validated against CCSD(T) data and the G0W0 approach of many-body perturbation theory. The accuracy of LDA-1/2 and G0W0 is found to be the same, where the latter is computationally much more involved. To get insight into the benefits and limitations of the LDA-1/2 method, we analyze the impact of each assumption made in deriving the methodology. Read more... |
|
W. Aggoune, C. Cocchi, D. Nabok, K. Rezouali, M. A. Belkhir, and C. Draxl With the example of hexagonal boron nitride, we demonstrate how the character of electron-hole (e−h) pairs in van der Waals bound low-dimensional systems is driven by layer stacking. Four types of excitons appear, with either a two- or three-dimensional spatial extension. Electron and hole distributions are either overlapping or exhibit a charge-transfer nature. We discuss under which structural and symmetry conditions they appear and they are either dark or bright. This analysis provides the key elements to identify, predict, and possibly tailor the character of e−h pairs in van der Waals materials. Read more... |
|
|
F. Caruso, D. Novko, and C. Draxl We investigate the effects of crystal lattice vibrations on the dispersion of plasmons. The loss function of the homogeneous electron gas (HEG) in two and three dimensions is evaluated numerically in the presence of electronic coupling to an optical phonon mode. Our calculations are based on many-body perturbation theory for the dielectric function as formulated by the Hedin-Baym equations in the Fan-Migdal approximation. The coupling to phonons broadens the spectral signatures of plasmons in the electron-energy loss spectrum (EELS) and it induces the decay of plasmons on timescales shorter than 1 ps. Our results further reveal the formation of a kink in the plasmon dispersion of the two-dimensional HEG, which marks the onset of plasmon-phonon scattering. Overall, these features constitute a fingerprint of plasmon-phonon coupling in EELS of simple metals. It is shown that these effects may be accounted for by resorting to a simplified treatment of the electron-phonon interaction which is amenable to first-principles calculations. Read more... |
|
 |
C. Vorwerk, C. Hartmann, C. Cocchi, G. Sadoughi, S. Habisreutinger, R. Felix, R. Wilks, H. Snaith, M. Bär, and C. Draxl In a combined theoretical and experimental work, we investigate X-ray absorption near-edge structure spectroscopy of the I L3 and the Pb M5 edges of the methylammonium lead iodide (MAPbI3) hybrid inorganic–organic perovskite and its binary phase PbI2. The absorption onsets are dominated by bound excitons with sizable binding energies of a few hundred millielectronvolts and pronounced anisotropy. The spectra of both materials exhibit remarkable similarities, suggesting that the fingerprints of core excitations in MAPbI3 are essentially given by its inorganic component, with negligible influence from the organic groups. The theoretical analysis complementing experimental observations provides the conceptual insights required for a full characterization of this complex material. Read more... |
|
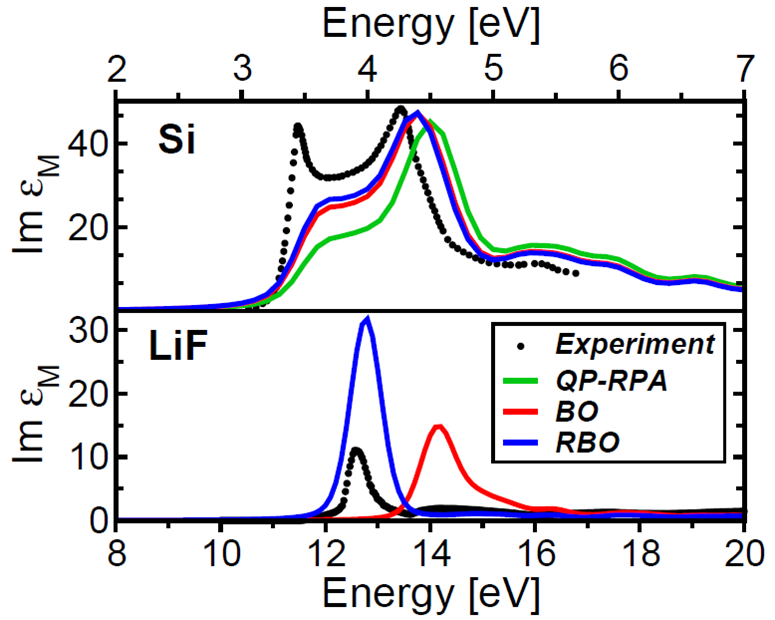
J. Gesenhues, D. Nabok, M. Rohlfing, and C. Draxl The power of the GW formalism is, to a large extent, based on the explicit treatment of dynamical correlations in the self-energy. This dynamics is taken into account by calculating the energy dependence of the screened Coulomb interaction W, followed by a convolution with the Green's function G. In order to obtain the energy dependence of W the prevalent methods are plasmon-pole models and numerical integration techniques. In this paper, we discuss an alternative approach, in which the energy-dependent screening is calculated by determining the resolvent, which is set up from a matrix representation of the dielectric function. On the one hand, this refrains from a numerical energy convolution and allows one to actually write down the energy dependence of W explicitly (like in the plasmon-pole models). On the other hand, the method is at least as accurate as the numerical approaches due to its multipole nature. We discuss the theoretical setup in some detail, give insight into the computational aspects, and present results for Si, C, GaAs, and LiF. Finally, we argue that the analytic representability is not only useful for educational purposes but may also be of avail for the development of theory that goes beyond GW. Read more... |
|
|
C. Vorwerk, C. Cocchi, and C. Draxl We present an ab initio study of core excitations of solid-state materials focusing on the role of electron-hole correlation. In the framework of an all-electron implementation of many-body perturbation theory into the exciting code, we investigate three different absorption edges of three materials, spanning a broad energy window, with transition energies between a few hundred to thousands of eV. Specifically, we consider excitations from the Ti K edge in rutile and anatase TiO2, from the Pb M4 edge in PbI2, and from the Ca L2,3 edge in CaO. We show that the electron-hole attraction rules x-ray absorption for deep core states when local fields play a minor role. On the other hand, the local-field effects introduced by the exchange interaction between the excited electron and the hole dominate excitation processes from shallower core levels, separated by a spin-orbit splitting of a few eV. Our approach yields absorption spectra in good agreement with available experimental data and allows for an in-depth analysis of the results, revealing the electronic contributions to the excitations, as well as their spatial distribution. Read more... |
|
 |
W. Aggoune, C. Cocchi, D. Nabok, K. Rezouali, M. Belkhir, and C. Draxl The optoelectronic properties of graphene/hexagonal boron nitride (h-BN) heterostructure demonstrate how a nanostructured combination of these materials can lead to a dramatic enhancement of light–matter interaction and give rise to unique excitations. In the framework of ab initio many-body perturbation theory, we show that such heterostructures absorb light over a broad frequency range, from the near-infrared to the ultraviolet (UV), and that each spectral region is characterized by a specific type of excitations. Besides the features, characteristic of the pristine constituents, charge-transfer excitations appear across the visible region. Remarkably, the spatial distribution of the electron and the hole can be selectively tuned by modulating the stacking arrangement of the individual building blocks. Our results open up unprecedented perspectives in view of designing van der Waals heterostructures with tailored optoelectronic features. Read more... |
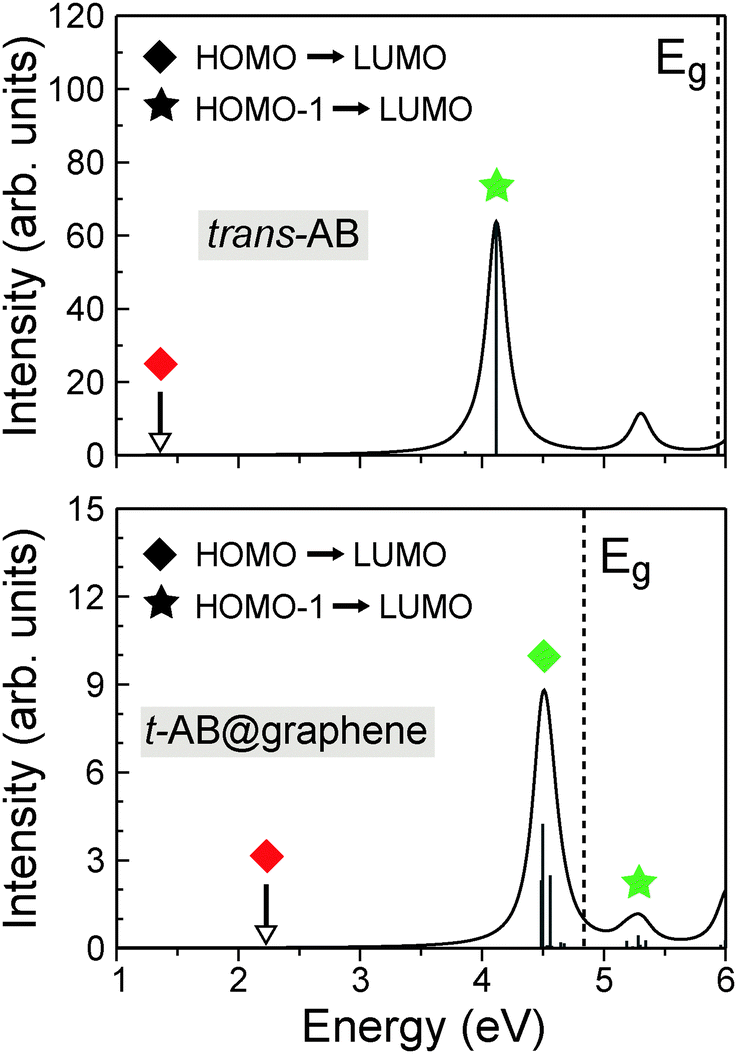 |
Q. Fu, C. Cocchi, D. Nabok, A. Gulans, and C. Draxl The impact of graphene on the photo-absorption properties of trans- and cis-azobenzene monolayers is studied in the framework of density-functional theory and many-body perturbation theory. We find that, despite the weak hybridization between the electronic bands of graphene and those of the azobenzene monolayers, graphene remarkably modulates the absorption spectra of the adsorbates. The excitation energies are affected via two counteracting mechanisms: substrate polarization reduces the band-gap of azobenzene, and enhanced dielectric screening weakens the attractive interaction between electrons and holes. The competition between these two effects gives rise to an overall blueshift of peaks stemming from intramolecular excitations, and a redshift of peaks from intermolecular ones. Even more interesting is that excitations corresponding to intermolecular electron–hole pairs, which are dark in the isolated monolayers, are activated by the graphene substrate. Our results demonstrate that the photoisomerization process of weakly adsorbed azobenzene undergoes notable changes on a carbon-based substrate. Read more... |
|
U. Hohenester and C. Draxl Gap plasmonics deals with the properties of surface plasmons in the narrow region between two metallic nanoparticles forming the gap. For subnanometer gap distances, electrons can tunnel between the nanoparticles, leading to the emergence of novel charge-transfer plasmons. These are conveniently described within the quantum corrected model by introducing an artificial material with a tunnel conductivity inside the gap region. Here we develop a methodology for computing such tunnel conductivities within the first-principles framework of density functional theory and apply our approach to a jellium model representative for sodium. We show that the frequency dependence of the tunnel conductivity at infrared and optical frequencies can be significantly more complicated than previously thought. Read more... |
|
|
C. Cocchi, H. Zschiesche, D. Nabok, A. Mogilatenko, M. Albrecht, Z. Galazka, H. Kirmse, C. Draxl, and C. T. Koch We present a joint theoretical and experimental study on core-level excitations from the oxygen K edge of β-Ga2O3. A detailed analysis of the electronic structure reveals the importance of O-Ga hybridization effects in the conduction region. The spectrum from O 1 s core electrons is dominated by excitonic effects, which overall redshift the absorption onset by 0.5 eV, and significantly redistribute the intensity to lower energies. Analysis of the spectra obtained within many-body perturbation theory reveals atomic fingerprints of the inequivalent O atoms. From the comparison of energy-loss near-edge fine-structure (ELNES) spectra computed with respect to different crystal planes, with measurements recorded under the corresponding diffraction conditions, we show how the spectral contributions of specific O atoms can be enhanced while quenching others. These results suggest ELNES, combined with ab initio many-body theory, as a very powerful technique to characterize complex systems, with sensitivity to individual atomic species and to their local environment. Read more... |
|
|
D. Nabok, A. Gulans, and C. Draxl The GW approach of many-body perturbation theory has become a common tool for calculating the electronic structure of materials. However, with increasing number of published results, discrepancies between the values obtained by different methods and codes become more and more apparent. For a test set of small- and wide-gap semiconductors, we demonstrate how to reach the numerically best electronic structure within the framework of the full-potential linearized augmented plane-wave (FLAPW) method. We first evaluate the impact of local orbitals in the Kohn-Sham eigenvalue spectrum of the underlying starting point. The role of the basis-set quality is then further analyzed when calculating the G0W0 quasiparticle energies. Our results, computed with the exciting code, are compared to those obtained using the projector-augmented plane-wave formalism, finding overall good agreement between both methods. We also provide data produced with a typical FLAPW basis set as a benchmark for other G0W0 implementations. Read more... |
|
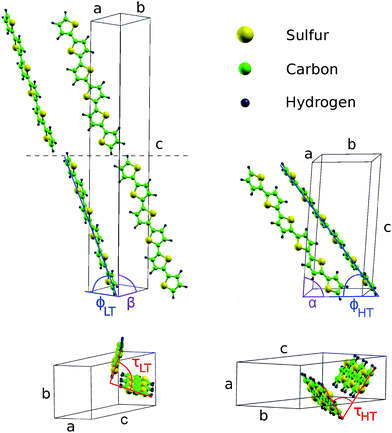 |
B. Klett, C. Cocchi, L. Pithan, S. Kowarik, and C. Draxl We present a joint theoretical and experimental study to investigate polymorphism in α-sexithiophene (6T) crystals. By means of density-functional theory calculations, we clarify that the low-temperature phase is favorable over the high-temperature one, with higher relative stability up to 50 meV per molecule. This result is in agreement with our thermal desorption measurements. We also propose a transition path between the high- and low-temperature 6T polymorphs, estimating an upper bound for the energy barrier of about 1 eV per molecule. The analysis of the electronic properties of the investigated 6T crystal structures complements our study. Read more... |
 |
Q. Fu, D. Nabok, and C. Draxl The two-dimensional semiconductors graphene fluoride and boron nitride are representative wide-bandgap components for van der Waals (vdW) heterostructures. We adopt their interface to investigate the impact of polarization effects on band gap and level alignment. We find a notable polarization-induced reduction of the materials’ band gap by 250 meV that we analyze in terms of an image-potential model. This value provides a lower limit of band-gap renormalization in 2D systems caused by polarization effects, and demonstrates the importance of many-body perturbation theory for a reliable prediction of energy-level alignment in 2D vdW heterojunctions. Read more... |
 |
C. Vorwerk, C. Cocchi, and C. Draxl We present Layer Optics, a versatile and user-friendly implementation, based on the solution of the Maxwell’s equations for anisotropic materials, to compute optical coefficients in anisotropic layered materials. We apply this tool for post-processing full dielectric tensors of molecular materials, including excitonic effects, as computed from many-body perturbation theory using the exciting code. For prototypical examples, ranging from optical to X-ray frequencies, we show the importance of combining accurate ab initio methods to obtain dielectric tensors, with the solution of Maxwell’s equations to compute optical coefficients accounting for optical anisotropy of layered systems. Read more... |
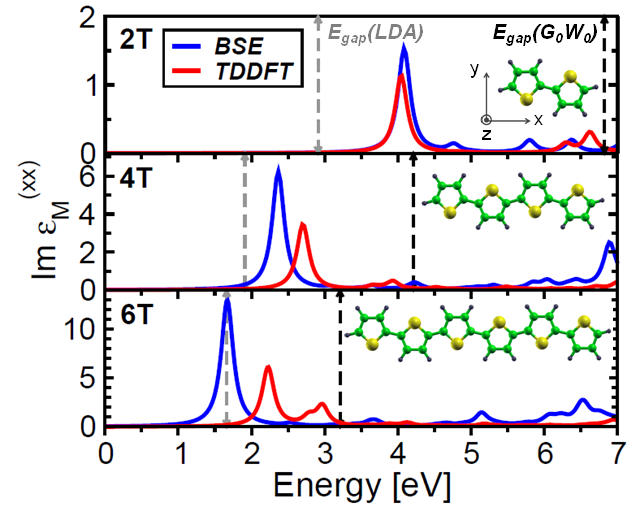 |
C. Cocchi and C. Draxl Time-dependent density-functional theory (TDDFT) is successful in describing excitation energies of finite systems, already in its most simple form, the adiabatic local-density approximation (ALDA). By confronting TDDFT with many-body perturbation theory, we clarify when and why this method can be trusted for molecular materials, where many-body effects can be crucial. We show that ALDA provides accurate results for excitations with mainly single-particle character. Conversely, when electron-hole correlations as well as local-field effects become decisive, only a many-body approach can quantitatively predict absorption features. Read more... |
 |
S.-A. Savu, G. Biddau, L. Pardini, R. Bula, H. F. Bettinger, C. Draxl, T. Chassé, and M. B. Casu Although physisorption is a widely occurring mechanism of bonding at the organic/metal interface, contradictory interpretations of this phenomenon are often reported. Photoemission and X-ray absorption spectroscopy investigations of nanorods of a substituted pentacene, 2,3,9,10-tetrafluoropentacene, deposited on gold single crystals reveal to be fundamental to identify the bonding mechanisms. We find fingerprints of a fractional charge transfer from the clean metal substrate to the physisorbed molecules. This phenomenon is unambiguously recognizable by a nonrigid shift of the core-level main lines while the occupied states at the interface stay mostly unperturbed, and the unoccupied states experience pronounced changes. The experimental results are corroborated by first-principles calculations. Read more... |
 |
S. Rigamonti, S. Botti, V. Veniard, C. Draxl, L. Reining, and F. Sottile A major obstacle for computing optical spectra of solids is the lack of reliable approximations for capturing excitonic effects within time-dependent density-functional theory. We show that the trustful prediction of strongly bound electron-hole pairs within this framework using simple approximations is still a challenge and that available promising results have to be revisited. Deriving a set of analytical formula we analyze and explain the difficulties. We deduce an alternative approximation from an iterative scheme guided by previously available knowledge, significantly improving the description of exciton binding energies. Finally, we show how one can "read" exciton binding energies from spectra determined in the random phase approximation, without any further calculation. Read more... |
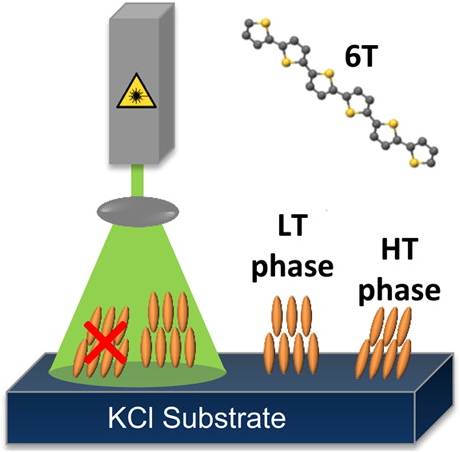 |
L. Pithan, C. Cocchi, H. Zschiesche, C. Weber, A. Zykov, S. Bommel, S.J. Leake, P. Schäfer, C. Draxl, S.M. Kowarik We demonstrate that the bimodal growth behavior of sexithiophene can be influenced strongly by light. α-Sexithiophene films grown in the dark on KCL consist of high- and low-temperature polymorphs, whereas in films grown under laser illumination the high temperature phase is significantly suppressed. This shows that light can act as a new control parameter in growth of organic semiconductors. Read more... |
 |
C. Draxl, D. Nabok, and K. Hannewald We address the question to what extent we can quantitatively describe hybrid materials and where we even miss a qualitative description. We argue when and why, for example, standard DFT may fall short when it comes to the electronic structure of organic/metal interfaces or where the framework of MBPT can or must take over. Selected examples of organic/inorganic interfaces, structural properties, electronic bands, optical excitation spectra, and charge-transport properties as obtained from DFT and MBPT highlight which properties can be reliably computed for such materials. Read more... |
People currently involved:
Olga Turkina (PhD student)
Sven Lubeck (PhD student)
Claudia Draxl (PI)
Dmitrii Nabok (postdoc)
Fabio Caruso (postdoc)
Pasquale Pavone (postdoc)
Andris Gulans (postdoc)
Santiago Rigamonti (postdoc)