News
People currently working in our group:






























Molecular Switches
Excitations of molecular
switches on surfaces: Insight from first principles
(2013-2017)
This project was embedded into the collaborative research center Elementary Processes in Molecular Switches at Surfaces, SFB 658, funded by the DFG.
By means of density-functional theory and many-body perturbation theory we investigate the interaction between molecular switches and different surfaces regarding structure, level alignment as well as optical excitations. In close collaboration with experimentalists, we address the interplay between intermolecular forces and molecule-substrate interaction, with the aim to understand the mechanisms driving the excitonic coupling of the molecules and the role of the surface in their switching behavior.
Selected Publications
Q. Fu, C. Cocchi, D. Nabok, A. Gulans, and C. Draxl
Graphene-modulated photo-absorption on adsorbed azobenzene monolayers
Phys. Chem. Chem. Phys. 19, 6196-6205 (2017).The impact of graphene on the photo-absorption properties of trans- and cis-azobenzene monolayers is studied in the framework of density-functional and many-body perturbation theory. Despite the weak electronic hybridization between graphene the azobenzene monolayers, graphene remarkably modulates the absorption spectra of the adsorbate. The excitation energies are affected via (i) polarization that reduces the band-gap of azobenzene, and (ii) enhanced dielectric screening that weakens the attractive interaction between electrons and holes. The competition between these two effects gives rise to an overall blueshift of peaks stemming from intramolecular excitations, and a redshift of peaks from intermolecular ones. Excitations corresponding to intermolecular electron–hole pairs, which are dark in the isolated monolayers, are activated by the graphene substrate. Our results demonstrate that the photoisomerization process of weakly adsorbed azobenzene undergoes notable changes on a carbon-based substrate.
C. Cocchi, T. Moldt, C. Gahl, M. Weinelt, and C. Draxl
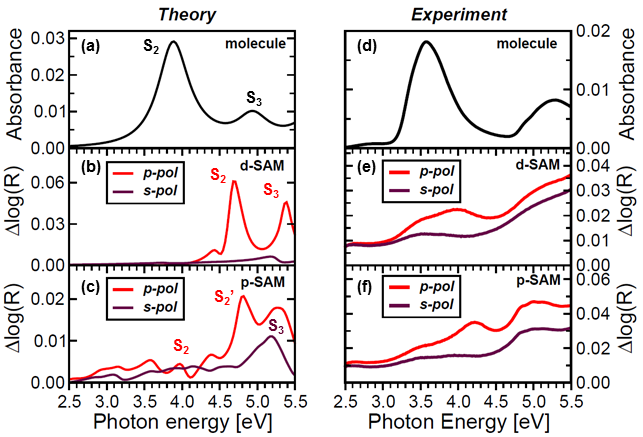
Optical properties of azobenzene-functionalized self-asselbled monolayers: Intermolecular coupling and many-body effects
J. Chem. Phys. 145, 234701 (2016).In a joint theoretical and experimental work the optical properties of azobenzene-functionalized self-assembled monolayers are studied at different molecular packing densities. While the optical excitations are intrinsically excitonic in nature, regardless of the molecular concentration, in densely-packed SAMs intermolecular coupling and local-field effects are responsible for a sizable weakening of the exciton binding strength. We show that distinct excitations involved in the photo-isomerization at low molecular concentrations are dramatically broadened by intermolecular interactions. Spectral shifts in the calculated DR spectra are in good agreement with the experimental results.
C. Cocchi and C. Draxl
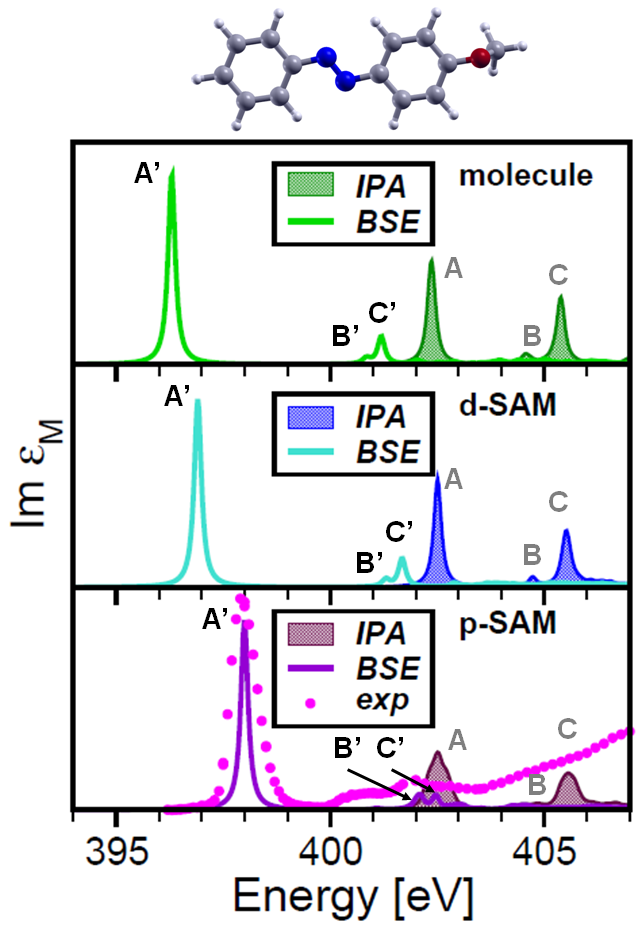
Bound excitons and many-body effects in X-ray absorption spectra of azobenzene-functionalized self-assembled monolayers
Phys. Rev. B 92, 205105 (2015).We study x-ray absorption spectra of azobenzene-functionalized self-assembled monolayers (SAMs), investigating excitations from the nitrogen K edge. Azobenzene with H-termination and functionalized with CF3 groups is considered. The Bethe-Salpeter equation is employed to compute the spectra, including excitonic effects, and to determine the character of the near-edge resonances. Our results indicate that core-edge excitations are intense and strongly bound: Their binding energies range from about 6 to 4 eV, going from isolated molecules to densely-packed SAMs. Electron-hole correlation rules these excitations, while the exchange interaction plays a negligible role.
C. Vorwerk, C. Cocchi, and C. Draxl
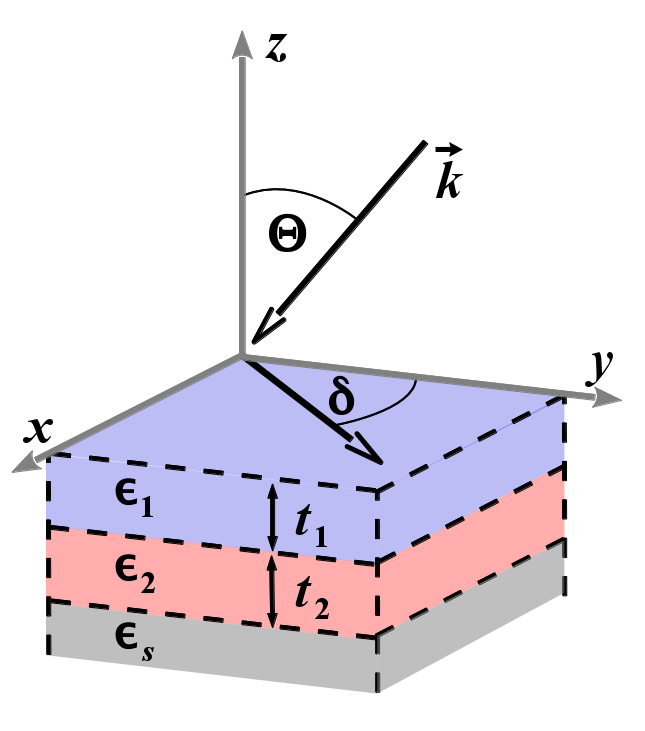
LayerOptics: Macroscopic modelling of optical coefficients in layered materials
Comp. Phys. Comm. 201, 119 (2016).Theoretical spectroscopy is a powerful tool to describe and predict optical properties of materials. While nowadays routinely performed, first-principles calculations only provide bulk dielectric tensors in Cartesian coordinates. These outputs are hardly comparable with experimental data, which are typically given by macroscopic quantities, crucially depending on the laboratory setup. Even more serious discrepancies can arise for anisotropic materials, e.g., organic crystals, where off-diagonal elements of the dielectric tensor can significantly contribute to the spectral features. Here, we present LayerOptics, a versatile and user-friendly implementation, based on the solution of the Maxwell’s equations for anisotropic materials, to compute optical coefficients in anisotropic layered materials. We apply this tool for post-processing full dielectric tensors of molecular materials, including excitonic effects, as computed from many-body perturbation theory using the exciting code.
People involved:
Qiang Fu (postdoc)
Benjamin Aurich (student assistant)
Caterina Cocchi (postdoc)
Albin Hertrich (master student)
Christian Vorwerk (master student)Philipp Häffner (bachelor thesis 2014)
Partner groups:
M. Weinelt / C. Gahl, B2, FU Berlin
P. Saalfrank / T. Klamroth, C2, Uni Potsdam